Introducción a la hemofilia con inhibidores
Los inhibidores son anticuerpos de tipo inmunoglobulina G (IgG) que neutralizan a los concentrados de factor de coagulación, reduciendo la eficacia del tratamiento. Se trata pues de una respuesta inmunológica contra el factor de coagulación administrado. No aparecen en todos los pacientes, y se considera la complicación más grave de la hemofilia ya que impiden al factor de coagulación funcionar correctamente y frenar la hemorragia1,2,3.
¿Cuál es el riesgo de desarrollar inhibidores?
La aparición de inhibidores es más frecuente en personas con hemofilia A que en hemofilia B. El riesgo de desarrollo de inhibidores es mucho mayor en hemofilia grave (entre el 20 y 30% de pacientes) que en hemofilia moderada o leve (5-10%)2.
El periodo de mayor riesgo de desarrollo de inhibidores es durante las primeras 50-75 infusiones de factor3,4.
Existen diferentes factores genéticos (ej. historia familiar de inhibidores, defectos graves en el gen del factor de coagulación, ascendencia africana…) y no genéticos (periodos de tratamiento intensivos, dosis de tratamiento, primeras exposiciones al factor…) que pueden contribuir a un riesgo mayor de presentar inhibidores 1,4.
Se calcula que hasta el 50% de los pacientes con hemofilia B e inhibidores frente al factor IX presentan riesgo de experimentar una grave reacción alérgica, llamada anafilaxis. Por ello es muy importante recibir este tratamiento en un centro especializado, en especial durante los primeros 20 tratamientos.

¿Cuándo sospechar la aparición de inhibidores?
En caso de desarrollarse inhibidores, estos suelen aparecen durante las primeras infusiones del factor de coagulación, y se debe sospechar en cualquier paciente que no responde al tratamiento, sobre todo si antes había respondido al mismo.
El hecho de presentar inhibidores no varía el lugar de la hemorragia, lo único que provoca es que el control de la hemorragia sea mucho más difícil.

¿Cómo se diagnostica la aparición de inhibidores?
La presencia de inhibidores siempre debe confirmarse mediante pruebas de laboratorio. Es recomendable hacer cada cierto tiempo una determinación de inhibidores en personas con hemofilia. Esto se realiza por un análisis en el laboratorio en el que se determina el tiempo que tarda la sangre en coagular mediante una prueba analítica rutinaria llamada “ensayo de tiempo de tromboplastina parcial activado (TTPA)”. La confirmación de la presencia de un inhibidor y la cuantificación del título se realiza en el laboratorio, preferentemente utilizando la prueba de Bethesda con la modificación de Nijmegen3.
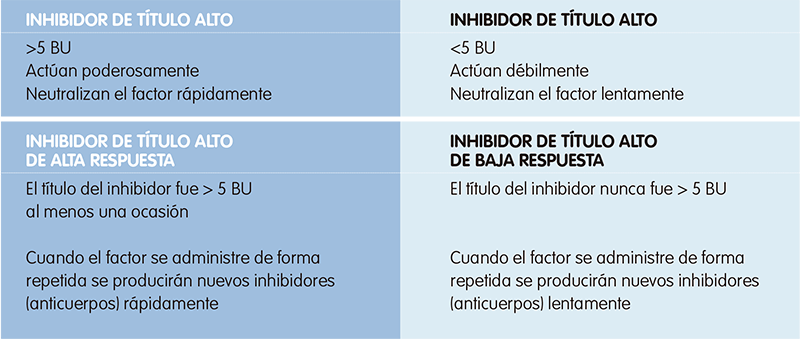
¿Cómo se clasifican los inhibidores?
La concentración de inhibidores varía de una persona a otra y puede variar a lo largo del tiempo en una misma persona. La cantidad de inhibidores en sangre se mide en unidades Bethesda (UB), que determinan la fuerza del inhibidor. De forma que4:
- Si las UB son superiores a 5 significa que los inhibidores neutralizan rápidamente el factor de coagulación (inhibidores de “título alto”)4.
- Si las UB son menores de 5 significa que los inhibidores son más débiles y neutralizan más lentamente al factor de coagulación (inhibidores de “título bajo”)4.
Además, también se clasifican en función de la respuesta de nuestro sistema inmunitario al factor de coagulación administrado en base a una exposición anterior al factor.
- Un inhibidor de “alta respuesta” es aquel que ha producido al menos en una ocasión una respuesta potente, mayor de 5 UB, y que al repetir la exposición al concentrado de factor producirá rápidamente nuevos inhibidores (alrededor de siete días)4.
- Un inhibidor de “baja respuesta” es aquel cuyo título en exposiciones previas al concentrado de factor siempre fue menor de 5 UB y producirá una respuesta inhibidora más débil frente al factor de coagulación4.
¿Cómo se trata la hemofilia con inhibidores?
Cuando se desarrollan inhibidores contra el factor VIII o factor IX, el proceso de coagulación también se puede poner en marcha con la administración de uno o varios factores que actúan a distintos niveles de la cascada de la coagulación, favoreciendo la formación del coágulo al bypasear la actividad del factor carente (agentes bypass)5.
Estos agentes bypass o agentes puentes, lo que hacen es participar en la formación del coágulo de forma distinta a la que lo hace el factor VIII. Estos agentes no remplazan directamente al factor de coagulación deficitario (que es frente al que se forman los inhibidores). De esta forma, los inhibidores no atacan a estos agentes5.
Otra alternativa en el abordaje de la hemofilia A con inhibidores es el uso de la terapia no sustitutiva, como puede ser un anticuerpo biespecífico que reconozca al factor IXa y al X6.


Dr. Víctor Jiménez Yuste
Jefe de Servicio de Hemostasia
Hospital Universitario La Paz, Madrid
¿Qué importancia considera que tiene la inmunotolerancia (ITI) para los pacientes con inhibidor?
La inmunotolerancia tiene como fin borrar un inhibidor desarrollado y permite que el paciente vuelva a
tener acceso a una profilaxis para el tratamiento de su enfermedad, siendo fundamental.
Referencias
- P. M. MANNUCCI, Q. SHI, S. BONANAD and R. KLAMROTH. Novel investigations on theprotective role of the FVIII/VWF complex in inhibitor development. Haemophilia. 2014;20 Suppl 6:2-16. doi: 10.1111/hae.12465.
- M. MAURO, E. BONETTI, R. BALTER et al. Recurrent episodes of anaphylaxis in a patient with haemophilia B: a case report. Blood Transfus.2016; 14: 582-4 DOI 10.2450/2016.0297- 15.
- C.L. KEMPTON and G. C. WHITE. How we treat a hemophilia A patient with a factor VIII inhibitor. Blood Transfus. 2009; 113: 11-17.
- K. GOMEZ, R. KLAMROTH, J. MAHLANGU et al. Key issues in inhibitor management in patients with haemophilia. Blood Transfus. 2014; 12 Suppl 1: s319-29 DOI 10.2450/2013.0246-12.
- Sociedad Española de Trombosis y Homeostasia. XI Curso de formación continuada. Trombosis y Hemostasia. Aran Ediciones S.L 2017
- Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nature Med 2012; 18: 1570–74
- S.J. SCHEP et al. Review of immune tolerance induction in hemophilia A. Blood Rev. 2018; 32; 4: 326-338