Introducción a la hemofilia
La hemofilia es un trastorno hemorrágico que afecta a la correcta coagulación de la sangre. Está causada por la baja o nula cantidad de un factor de coagulación que dificulta o impide la formación del coágulo de fibrina que permite controlar el sangrado1.
Los factores de coagulación son 13 proteínas (factores) que están relacionadas entre sí y forman la “cascada de la coagulación”, cuya función es formar el coágulo. En condiciones normales, la coagulación de la sangre impide la aparición de hemorragias y ayuda a que los sangrados cedan al poco tiempo de que se haya producido una lesión vascular.
Las personas con hemofilia no sangran más rápido de lo normal, pero sí pueden tener un sangrado más prolongado debido a la cantidad insuficiente del factor de coagulación. De esta forma, las personas con esta enfermedad pueden tener hemorragias importantes provocadas por traumatismos pequeños.
En función de la deficiencia de factor de coagulación que tenga una persona, existen distintos tipos de hemofilia, siendo los más frecuentes1:
- Hemofilia A, en la que hay un déficit en el factor VIII de la cascada de coagulación.
- Hemofilia B, en la que no hay suficiente factor IX de la cascada de coagulación.
¿Qué es la cascada de la coagulación?
Cuando un vaso se lesiona se contrae para limitar la cantidad de sangre en el área lesionada, y las plaquetas se adhieren al sitio de la lesión, liberando una serie de señales químicas que atraen a otras células, aglutinándose para formar el tapón plaquetario.
En la superficie de estas plaquetas, que ahora están activadas, existen distintos factores de la coagulación que se activan debido a la acción de otro factor que se ha activado previamente, de forma que un factor activa a otro siguiendo un orden. Al final de este proceso, se forma el coágulo de fibrina que detiene el sangrado. Esta compleja reacción se conoce como cascada de la coagulación.
Niveles de gravedad de la hemofilia
La gravedad de la hemofilia se ha diferenciado en tres niveles en función de la concentración del factor de coagulación existente en el plasma sanguíneo, teniendo en cuenta que el nivel de los factores de coagulación VIII y IX en una persona no afectada varía entre el 50% y el 150%.
De esta forma la gravedad se clasifica según la siguiente tabla1:

¿Cuántas personas padecen hemofilia?
Según los datos del año 2012 de la Federación Mundial de Hemofilia, se estima que la enfermedad aparece en uno de cada 10.000 nacimientos, y se calcula que en todo el mundo hay aproximadamente 400.000 personas con hemofilia1.
Los datos disponibles para la población española correspondientes al año 2009 tenían registrados 3.000 pacientes con hemofilia, de los cuales el 87% presentaban hemofilia A, el 13% hemofilia B y había un 3% de hemofílicos en los que no se había determinado el tipo de enfermedad. Respecto a la gravedad de la enfermedad, el 51,4% de los pacientes estaban catalogados de grado leve, 16,3% de moderada y 32,1% de grave mientras que el 0,2% era de origen desconocido2. Por rangos de edad, los pacientes entre 25 y 49 años eran los más numerosos en ambos tipos de hemofilia2.

¿Cómo se diagnostica la hemofilia?
Normalmente el diagnóstico es ágil cuando existen antecedentes familiares de la enfermedad. En caso de que estos no existan, puede existir sospecha diagnóstica de hemofilia en personas que presenten hemorragias espontáneas o ante hemorragia importantes producidas por traumatismos leves. Las manifestaciones de la hemofilia son muy similares tanto en el tipo A como en el tipo B1.
Para el diagnóstico de la enfermedad se debe realizar un análisis de sangre para comprobar si aparecen alteraciones en las pruebas de coagulación, y además se evidenciará la deficiencia del factor de la coagulación deficitario. También se puede realizar un diagnóstico molecular mediante la identificación de la alteración genética en el gen codificante del factor1.
¿Cómo se transmite la enfermedad?
La hemofilia es una enfermedad hereditaria recesiva que está ligada al cromosoma X.
Los cromosomas son estructuras (ADN y proteínas) que contienen la información que determina nuestras características. Nuestro organismo tiene 23 parejas de cromosomas; de cada pareja, un cromosoma es heredado del padre y el otro de la madre. De estas 23 parejas, el par 23 es el encargado de determinar el sexo. Son los llamados cromosomas sexuales. Existen dos cromosomas sexuales (X e Y): las mujeres son XX, y los varonesXY.
Los genes afectados en la hemofilia A y B están en el cromosoma X. En las mujeres, aunque uno de sus dos cromosomas X esté alterado (y pueden transmitirlo a sus hijos), si el otro es normal, lo más probable es que la mujer no presente síntomas. Estas mujeres se denominan portadoras asintomáticas. En los hombres, cuando el cromosoma X está afectado es suficiente para causar la enfermedad. Es por eso que la enfermedad la padecen más los hombres.
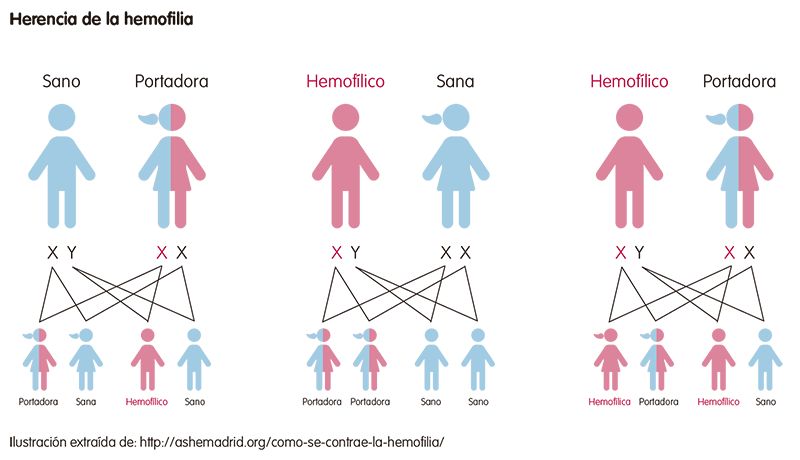
Esto determina que la hemofilia afecte principalmente a los varones, aunque de forma poco frecuente también la pueden presentar las mujeres, cuando el padre tiene la hemofilia y la madre es portadora.
La alteración genética causante de la hemofilia puede ocurrir también de forma espontánea (“de novo”) representando el 30% de los casos de hemofilia.
¿Cómo se manifiesta la hemofilia?
La principal manifestación clínica de la hemofilia es la tendencia que tienen los pacientes a los sangrados, siendo más duraderos que en una persona no hemofílica.
En algunos niños con hemofilia grave, los sangrados pueden no aparecer hasta que el niño empieza a caminar o a correr.
En los pacientes con hemofilia moderada, los sangrados pueden aparecer cuando exista un traumatismo o cuando se realice alguna intervención quirúrgica.
Los pacientes con hemofilia leve raramente presentan sangrados espontáneos, aunque sí pueden tener sangrados importantes a causa de traumatismos graves o bien, cuando se someten a cirugía.
La mayoría de los sangrados en pacientes con hemofilia son en las articulaciones y se denominan hemartros. Estos sangrados se presentan principalmente en las articulaciones bisagra (tobillos, rodillas y codos), en los músculos (sobre todo en los profundos como el psoas ilíaco, los músculos de la pantorrilla y los músculos del antebrazo) o en las mucosas como la boca, las encías, la nariz y el aparato urinario.

Sin embargo, a veces también pueden aparecer sangrados intracraneales, en el cuello, en la garganta o sangrados gastrointestinales que pueden poner en peligro la vida del paciente, y que precisan de tratamiento inmediato o urgente.
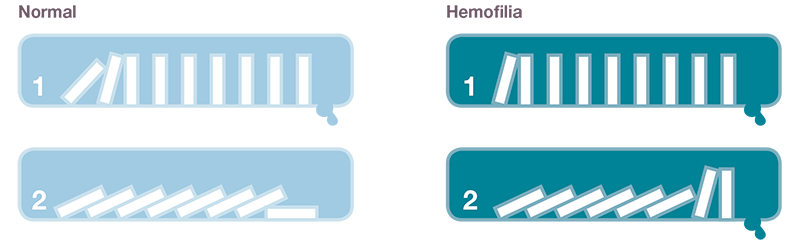
¿Qué es un hemartros?
El hemartros es una hemorragia en el interior de la articulación. Aproximadamente el 80-90% de los episodios de sangrado en pacientes hemofílicos ocurren en el sistema musculoesquelético. Apareciendo principalmente en las articulaciones que tienen tejido sinovial y por orden de frecuencia suelen suceder en codos, rodillas, tobillos, caderas y hombros. Habitualmente son consecuencia de un traumatismo, aunque en ocasiones aparecen espontáneamente. También se pueden ver afectados, aunque en menor medida, la cadera, el hombro y la muñeca1,2,3,11.
Los hemartros a su vez, afectan a la membrana sinovial de la articulación haciendo que ésta se inflame, proceso conocido como sinovitis. Cuando la membrana sinovial está afectada, tiene mayor tendencia a sangrar, generándose así un círculo vicioso (hemartrosis-sinovitis- hemartrosis). Todo este cuadro se puede agravar debido a la atrofia muscular producida por la inmovilidad y el dolor, que a su vez hace que la articulación sea más inestable y por tanto que tenga más riesgo de sangrar1,2.
Muchos pacientes describen el inicio de la hemorragia intra-articular como una sensación de hormigueo y cierta opresión en la articulación que ocurre antes de que aparezcan los signos clínicos de la hemartrosis. Esto es lo que se conoce como “aura”1.
Se conoce como articulación diana a la articulación en la que se producen 3 o más sangrados en un periodo de 6 meses1,3.
La característica del hemartros agudo es la pérdida rápida de movilidad asociada a dolor, hinchazón, calor en la piel en toda la zona articular e incapacidad funcional. Para disminuir el dolor, la articulación se encontrará en flexión1.
El hemartros recurrente se diferencia de la sinovitis en que el primero es de inicio agudo, se acompaña de dolor intenso o moderado, se adopta una posición en flexión de la articulación para evitar el dolor, la movilidad de la articulación está limitada, existe pérdida de fuerza articular y responde rápidamente al tratamiento con factores sustitutivos. Por su parte, la sinovitis tiene un inicio lento con poco dolor o sin él, con movilidad articular normal y no responde inmediatamente al tratamiento sustitutivo, pero si a los antiinflamatorios y/o a los corticoides1.
El hemartros y la sinovitis se pueden prevenir o reducir de forma importante con el tratamiento profiláctico con los factores de coagulación deficitarios, mejorando la calidad de vida del paciente y permitiendo una actividad física normal1.
¿Cómo se trata la hemofilia?
Introducción
El sangrado o la hemorragia es la manifestación clínica más importante de la hemofilia.
En los pacientes con hemofilia grave, ya sea A o B, los cuadros hemorrágicos varían y en algunos casos no aparecen hasta que el niño empieza a caminar y a caerse. En las formas graves, las hemorragias son habituales en músculos y en articulaciones1.
En los casos de hemofilia A o B moderada o leve, puede no haber sangrados hasta que se produzca un traumatismo o hasta el momento en el que se realice algún tipo de cirugía. La aparición de estos sangrados dependerá de los niveles de FVIII o FIX que tenga el paciente.
El tratamiento de la hemofilia consiste en la administración intravenosa del factor de coagulación deficitario.
El objetivo principal del tratamiento de la hemofilia es prevenir y tratar el sangrado cuando aparezca, así como sus posibles complicaciones y siempre que sea posible se hará con infusión del concentrado de factor específico deficitario1.

El tratamiento con factores de coagulación se conoce como tratamiento sustitutivo3. Existen dos formas o modalidades de tratamiento:

En personas con hemofilia moderada o leve, el tratamiento con factor de coagulación suele administrase sólo en caso de sangrados. Sin embargo, en personas con hemofilia grave, el factor de coagulación se suele administrar de forma regular (dos/tres veces por semana). Este tipo de tratamiento se denomina profilaxis. La profilaxis es el tratamiento ideal en pacientes con hemofilia grave, ya que puede ayudar a minimizar los sangrados y el daño a largo plazo en las articulaciones debido a sangrados internos repetidos1,3.
Tratamiento episódico o a demanda
Consiste en la administración intravenosa del factor de la coagulación deficitario (FVIII o FIX) para tratar los sangrados. En esta modalidad, el tratamiento sustitutivo se administra en el momento en que existe una hemorragia1. También se utiliza esta modalidad cuando se deben realizar exploraciones médicas que puedan causar sangrado, como las biopsias, y en caso de cirugía3.
El objetivo del tratamiento del hemartros agudo es parar la hemorragia tan pronto como sea posible, e idealmente debe realizarse lo antes posible cuando el paciente reconoce los signos del “aura”3 . Es frecuente que los pacientes describan estos signos de “aura” como una leve molestia, una sensación de cosquilleo y tirantez en la articulación que limita levemente el movimiento articular1,3.

Tratamiento Profiláctico o Profilaxis
El tratamiento profiláctico tiene como objetivo evitar o reducir lo sangrados, con el fin de impedir o retrasar el deterioro articular que se produce cuando las hemorragias son repetidas1,3. Esta modalidad proporciona la protección necesaria para prevenir la aparición de hemorragias espontáneas o secundarias a un traumatismo leve1.
La profilaxis reduce el número de hemartrosis y mejora el estado articular del paciente, ayudando a su integración laboral, deportiva y en las actividades de ocio, y de esta forma se mejora también la calidad de vida4.
La profilaxis se realiza con concentrados plasmáticos o recombinantes intravenosos administrados varias veces por semana. Han demostrado ser eficaces y seguros, aunque hasta en el 30% de los pacientes con hemofilia A grave se desarrolla inhibidor5.
Tratamiento no farmacológico
En caso de hemorragia aguda de la articulación (hemartros), se deberá administrar el tratamiento farmacológico prescrito por el médico, a ser posible en las primeras dos horas desde el inicio del sangrado. Después se aplicará la técnica “RICE” (por sus siglas en inglés) que significa4:
• Rest: Reposo
• Ice: Hielo
• Compress: Compresión
• Elevate: Elevación
Cuando se ha solucionado el proceso de sangrado agudo, se recomienda iniciar fisioterapia y/o rehabilitación personalizada.
Desde siempre, la rehabilitación y la fisioterapia han sido parte del tratamiento de las lesiones musculoesqueléticas, y ello es aplicable también a los pacientes con hemofilia.
Rehabilitación
Además del tratamiento farmacológico cuando existe artropatía hemofílica instaurada, la rehabilitación es necesaria para recuperar física, psíquica y laboralmente al paciente4.
La rehabilitación es importante para:
• Mantener la movilidad articular máxima posible (amplitud del movimiento), ya que esta movilidad influye en las actividades de la vida diaria y en la progresión del proceso degenerativo5.
• Mejorar el equilibrio, disminuyendo de esta forma el riesgo de caídas. En la rehabilitación (y en la fisioterapia) se debe trabajar no solo la articulación sino también las posibles alteraciones en los ligamentos que podrían provocar inestabilidad5.
• También se trabaja la fuerza muscular, ya que al mejorar ésta se reduce el riesgo de hemorragia.
En los pacientes que aún no presentan artropatía, se aconseja realizar ejercicios de movilización libre de todas las articulaciones y programas de potencia muscular. Es recomendable hacer más ejercicios isométricos que isotónicos4.
También se recomienda que se realice una rutina de actividad física o deportiva siendo de elección las actividades aeróbicas y de bajo impacto.
Cuando ya existe artropatía establecida, se recomienda realizar ejercicios de potenciación muscular de los grupos musculares relacionados con la articulación afectada. Es importante ser constante en la realización de los ejercicios, convirtiéndolos en una rutina4.
Fisioterapia
La fisioterapia en el paciente hemofílico tiene como objetivo el mantenimiento de la articulación lo más sana posible evitando deformidades, aliviar el dolor y reforzar la musculatura4,6.
Es recomendable que el fisioterapeuta esté especializado en el tratamiento de pacientes con hemofilia, ya que deberá realizar una evaluación completa del aparato locomotor y ajustar el programa de fisioterapia a las necesidades y al estilo de vida de cada paciente. Además, deberá realizar manipulaciones cuidadosas para evitar provocar daño, así como confirmar que el paciente se ha administrado el factor antes de la sesión4.
Existen distintos niveles de fisioterapia:
• Fisioterapia preventiva para mejorar la situación física y prevenir lesiones agudas. Es muy importante evitar el sobrepeso. Este tipo de fisioterapia se recomienda en niños en los que se realiza profilaxis y no presentan hemorragias o tienen muy pocas4.
• Fisioterapia terapéutica, que se hará después de un sangrado para aliviar el dolor, mejorar la movilidad y evitar la atrofia muscular fortaleciendo el músculo4.
• Fisioterapia de mantenimiento que se hace en los pacientes en los que la lesión articular ya está muy evolucionada. El objetivo es mantener en lo posible la movilidad de la articulación y evitar la atrofia4.
Una técnica de fisioterapia que se utiliza en el tratamiento de las hemorragias articulares (y también en el tratamiento de las sinovitis) es la crioterapia o terapia de frío. Se trata de aplicar frío localmente durante 10-15 minutos en la articulación afectada después de la lesión o del ejercicio que ha provocado un sobreesfuerzo. Esto se repite dos o tres veces al día4.
A pesar de los posibles problemas que supone el ejercicio, los beneficios del mismo superan las posibles complicaciones, siempre que se realice un ejercicio acorde con cada paciente y previa consulta con su médico. La práctica de ejercicio es beneficiosa para la condición física del paciente, el desarrollo neuromuscular y la calidad de vida.
¿Qué es la Farmacocinética (PK)?
La farmacocinética es el conjunto de procesos que sufre el fármaco a su paso por el organismo, o dicho de otra forma, lo que ocurre en el organismo al administrar un medicamento. El estudio de la farmacocinética nos ayuda a definir algunas características como la dosis del fármaco que queremos administrar o el intervalo entre dosis6.
Cuando se administra el fármaco (por ejemplo, el FVIII en hemofilia A) por medio de una infusión venosa, todo él llega a la sangre, se distribuye por el organismo y se metaboliza eliminándose lentamente. Cada persona lo elimina a una velocidad, por eso decimos que cada persona tiene su ritmo6,7.
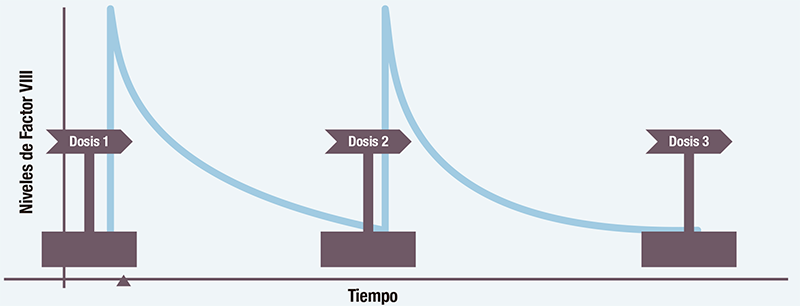
Generalmente, la farmacocinética se representa con una curva en la que los niveles de factor VIII van disminuyendo con el tiempo según el fármaco se va eliminando. Además, la curva de farmacocinética ayuda a conocer los niveles “pico” y “valle” de cada individuo8.
Conocer el tiempo en el que se alcanza el nivel mínimo de factor VIII permite determinar la frecuencia adecuada de las dosis, es decir cuándo se han de administrar las infusiones. De este modo se reduce al máximo el tiempo en los que los niveles de factor VIII se encuentran por debajo de los requeridos para asegurar la prevención adecuada de los sangrados en aquellos pacientes que están en profilaxis8.
Conocer el nivel máximo de factor VIII, así como el tiempo en el que se alcanza, es especialmente relevante para ajustar el tratamiento preventivo en situaciones de riesgo elevado de lesiones y sangrados, como operaciones o actividad física intensa.
Cada paciente tiene una curva específica ya que, metaboliza el factor VIII a una velocidad diferente. Esto explica por qué cada paciente responde de manera diferente al tratamiento y por qué es recomendable la personalización del mismo. Además, se pueden planificar las actividades a realizar según el nivel de factor del que disponga cada persona, según la eliminación que tenga8.
La Personalización del Tratamiento

Mª Teresa Álvarez Román
Especialista en Hematología y Hemoterapia
Hospital Universitario La Paz, Madrid
¿Qué significa para usted personalizar el tratamiento en hemofilia? ¿Qué beneficios tiene la profilaxis personalizada y qué aporta para usted y para sus pacientes?
Es adaptar el tratamiento del paciente a sus características. En el caso de la hemofilia, es muy importante para controlar la farmacocinética, la cantidad del producto infundido y favorece la adherencia a la profilaxis.
La determinación de la curva de PK en pacientes con Hemofilia A
La determinación de la curva farmacocinética del factor VIII es un factor muy importante a la hora de personalizar el tratamiento. Sin embargo, no es el único factor que debe tenerse en cuenta a la hora de personalizar el tratamiento. El médico debe considerar además los siguientes aspectos de cada paciente9,10.
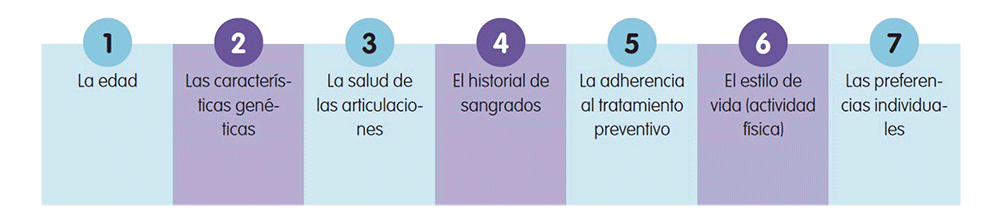
Todos estos criterios permiten seleccionar el tratamiento preventivo adecuado y alcanzar los mejores resultados. De este modo, se consigue minimizar la carga de la enfermedad.
Referencias
- A. SRIVASTAVA, A. K. BREWER, E. P. MAUSER-BUNSCHOTEN et al. Guidelines for the management of hemophilia. Haemophilia. 2013; 19(1). e1-47.
- W.D. JANSEN, G. ROOSENDAAL and P. J. G. LABEFER. Understanding haemophilic arthropathy: an exploration of current open issues. BJH, 2008; 143, 632–640.
- V.S. BLANCHETTE, N.S. KEY, L.R. LJUNG. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014; 12: 1935–1939.
- LÓPEZ-CABARCOS C, QUEROL F, MORENO S, CRESPO A, CUESTA R, ALONSO C, et al. Recomendaciones sobre rehabilitación en hemofilia y otras coagulopatías 2009. Comisión científica de la Real Fundación Victoria Eugenia. Ediciones de la Real Fundación Victoria Eugenia y Federación Española de Hemofilia
- C. ALTISENT, Mª T. ÁLVAREZ, D.B. TABOADA et al. Altas de hemofilia. Madrid: Momento Medico s.r.l.; 2013.
- SHAPIRO, AD. Et al. (2005). Use of pharmacokinetics in the coagulation factor treatment of patients with haemophilia. Haemophilia. 11. 571-582
- COLLINS, P. et al. (2011). Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. 17. 2-10.
- COLLINS, P.W. et al. (2010). Factor VIII requirement to maintain a target plasma level in the prophylactic treatment of severe hemophilia A: influences of variance in pharmacokinetics and treatment regimens. Journal of Thrombosis and Haemostasis. 8(2).269-75
- VALENTINO, L.A. (2014). Considerations in individualizing prophylaxis in patients with haemophilia A. Haemophilia. 20(5).607-15.
- OLDENBURG, J. (2015). Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 125(13).2038-44
- RODRIGUEZ-MERCHAN E.C et al. Joint Protection in Haemophilia. Haemophilia (2011), 17 (Suppl. 2), 1–23